Peer Reviewed
An Unexpected Outcome in an Adolescent With Juvenile Ankylosing Spondylitis
Answer: B. Fracture
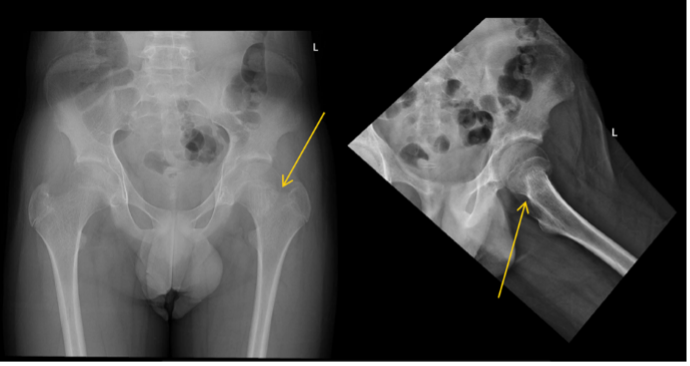
Given the patient’s previous vertebral fractures, low bone density, and recent trauma, fracture was suspected. Initial hip X-ray was reported by the radiology team as negative for signs of fracture. However, after review of the X-ray and the patient’s clinical condition by the pediatrician, a reread of the hip X-ray was requested. A suspected left femoral neck fracture was noted following the reread and the radiology team recommended magnetic resonance imaging (MRI) for confirmation (Figures 1 and 2), which ultimately confirmed the diagnosis.

Figure 1. Bilateral hip X-ray with mildly impacted left femoral neck fracture (yellow arrow).
Figure 2. MRI of the left hip with a nondisplaced left femoral neck fracture (yellow arrow), marked marrow edema of the proximal femur (red arrow), and joint effusion with synovitis (white arrow).
Severe pain with refusal to bear weight is usually caused by acute infections, malignancy, trauma, transient synovitis, or avascular necrosis. His recurrent right hip pain due to JAS put him at increased risk for similar symptoms in the contralateral hip; however, JAS pain is usually less severe with a more insidious onset causing a limp rather than inability to bear weight. He had no evidence of infection or malignancy. SCFE was possible given his age but was less likely since he didn’t hold his leg in external rotation and his BMI was within normal range. Transient synovitis could not be ruled out and was a diagnosis of exclusion.
Treatment and management. The patient underwent in situ 3 cannulated screw fixation and was placed on crutches for 6 weeks. He was to continue supplementation with 2000 IU of vitamin D, add a small amount of sun exposure, add 1300 mg of calcium in his diet, and reestablish with an endocrinologist.
Outcome and follow-up. He was mostly consistent with vitamin D supplementation but did not add sun exposure or measure calcium in his diet. The endocrinologist felt that his bones were very fragile and thin for his age, so a dual X-ray absorptiometry (DEXA) scan was completed, which showed a low lumbar spine bone mineral density (BMD) of 0.813 g/cm² and a Z score of -0.6. He was referred to a geneticist for evaluation of bone fragility syndromes.
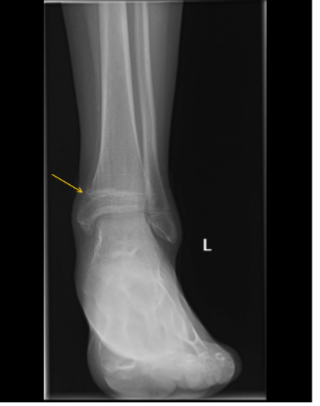
While awaiting evaluation, he sustained a Salter-Harris II fracture of the distal left tibia after tripping while using crutches (Figure 3).
Figure 3. X-ray of the left ankle with fracture through the medial tibial metaphysis and physis (yellow arrow) with periosteal reaction and osteopenia.
The genetics team evaluated for hypophosphatasia (HPP), osteogenesis imperfecta (OI), and other bone fragility syndromes. An alkaline phosphatase level within normal limits ruled out HPP. Although he did not have significant findings for OI, a mild form could not be ruled out. A bone fragility gene panel revealed a mutation in COL1A2 (c.964G>A [p.Gly322Ser]), a pathogenic variant for OI, and NOTCH2 (c.3467A>G [p.Asn1156Ser]), a variation of unknown significance.
His indomethacin was discontinued, vitamin D supplementation was increased to 4000 IU/day, and pamidronate infusions were started (0.5 mg/kg for initial dose, then 1 mg/kg every 4 months). Additional recommendations included annual DEXA scans and spine X-rays for scoliosis and fractures, hearing evaluation every 3-5 years, cervical X-rays prior to sports, and MRI of the skull base for basilar impression given any neurologic symptoms.
The patient has been without fractures for the past 8 months. His mom and sister both tested positive for the OI variant.
Discussion. JAS is a seronegative spondyloarthropathy strongly associated with HLA-B27. It is an inflammatory condition affecting the spine and large joints, especially the sacroiliac joints. Clinical features include joint pain, morning stiffness, and uveitis. Numerous studies have shown an increased risk of vertebral fractures in ankylosing spondylitis1-6; however, the incidence of nonvertebral fractures is less clear. One case-control study found no significant increased risk of forearm or hip fractures4, and another population-based cohort study found no increase in the risk of limb fractures.2
OI is a rare, heritable connective tissue disorder. Autosomal dominant mutations in the genes encoding type I collagen, COL1A1, and COL1A2, are the most common causes of OI, but at least 17 other associated genes have been identified.7 OI has multiple subtypes ranging from premature osteoporosis to perinatal death. Clinical features include low bone mass, bone fragility, blue or grey sclera, hearing impairment, dentinogenesis imperfecta, and spine deformities.7,8 While vertebral fractures can be associated with OI, evidence suggests that extremity fractures are more common. A retrospective study of pediatric patients diagnosed with OI found that the most common fractures at diagnosis were in the extremities.9 Another population-based cohort study found that boys with OI aged 0 to 19 years had the highest fracture rates in forearm, femur, lower leg, and ankle.10
In patients with multiple or suspicious fractures, genetic testing can be used to confirm the diagnosis. Type I collagen is a protein abundant in bone, skin, and other connective tissue that forms a triple helix with characteristic Gly-X-Y amino acid repeats that are necessary for structure and stability.11 Our patient had a mutation in the α2(I) chain of type I collagen with the usual glycine residue at position 322 being replaced by serine. This substitution disrupts protein function and is a pathologic variant of OI.12 He also had a mutation in NOTCH2, which encodes a transmembrane receptor protein that regulates gene expression.13 NOTCH2 mutations are associated with Hajdu-Cheney syndrome and are characterized by facial anomalies, osteoporosis, and short stature, as well as Alagille syndrome, which presents with severe renal disease, skeletal abnormalities, and hepatic cholestasis.13,14 The variant identified in our patient’s panel has not been found to significantly alter protein function.15 The lack of dysmorphic facial features, short stature, and renal or hepatic complications in our patient make the significance of this variant unknown and further support the diagnosis of OI.
Treatment of JAS includes NSAIDS, corticosteroids, and physical therapy.1,4,16 Conversely, bisphosphonate therapy has long been used to treat children with OI showing increases in BMD, reductions in fractures, and restoration of vertebral size and shape following compression fracture.7 Bisphosphonates, such as pamidronate and zoledronate, exert their therapeutic effects by inhibiting osteoclast function, thereby inhibiting bone resorption and promoting bone growth and strength.16 Treatment with pamidronate was chosen for our patient given his BMD within normal limits, mid-puberty status which has natural increases in BMD, and to avoid overtreatment as zoledronate is stronger.
Given overlapping symptoms, it is beneficial to review the features of these conditions. This case exemplifies the importance of obtaining a detailed history and physical examination, the value of continuity of care with a primary care physician, and the increasing utility of multi-gene screening for enhanced diagnostic evaluation.
References
1. Weiss RJ, Wick MC, Ackermann PW, Montgomery SM. Increased fracture risk in patients with rheumatic disorders and other fnflammatory diseases — a case-control study with 53,108 patients with fracture. J Rheumatol. 2010;37(11):2247-2250. doi:10.3899/jrheum.100363
2. Cooper C, Carbone L, Michet CJ, Atkinson EJ, O'Fallon WM, Melton LJ. Fracture risk in patients with ankylosing spondylitis: a population based study. J Rheumatol. 1994;21(10):1877-1882.
3. Mitra D, Elvins DM, Speden DJ, Collins AJ. The prevalence of vertebral fractures in mild ankylosing spondylitis and their relationship to bone mineral density. se 2000;39(1):85-89. doi:10.1093/rheumatology/39.1.85
4. Vosse D, Landewe R, van der Heijde D, van der Linden S, van Staa TP, Geusens P. Ankylosing spondylitis and the risk of fracture: results from a large primary care-based nested case-control study. Ann Rheum Dis. 2009;68(12):1839-1842. doi:10.1136/ard.2008.100503
5. Prieto-Alhambra D, Muñoz-Ortego J, De Vries F, et al. Ankylosing spondylitis confers substantially increased risk of clinical spine fractures: a nationwide case-control study. Osteoporos Int. 2015;26(1):85-91. doi:10.1007/s00198-014-2939-3
6. Sambrook PN, Geusens P. The epidemiology of osteoporosis and fractures in ankylosing spondylitis. The Adv Musculoskelet Dis. 2012;4(4):287-292. doi:10.1177/1759720x12441276
7. Palomo T, Vilaça T, Lazaretti-Castro M. Osteogenesis imperfecta: diagnosis and treatment. Curr Opin Endocrinol Diabetes Obes. 2017;24(6):381-388. doi:10.1097/med.0000000000000367
8. Weaver JS, Revels JW, Elifritz JM, Whitlow B, Retrouvey M, Wang SS. Clinical manifestations and medical imaging of osteogenesis imperfecta: fetal through adulthood. Acta Med Acad. 2021;50(2):277-291. doi:10.5644/ama2006-124.343
9. Greeley CS, Donaruma-Kwoh M, Vettimattam M, Lobo C, Williard C, Mazur L. Fractures at diagnosis in infants and children with osteogenesis imperfecta. J Pediatr Orthop. 2013;33(1):32-36. doi:10.1097/bpo.0b013e318279c55d
10. Folkestad L, Hald JD, Ersboll AK, et al. Fracture rates and fracture sites in patients with oosteogenesis imperfecta: a nationwide register-based cohort study. J Bone Miner Res. 2017;32(1):125-134. doi:10.1002/jbmr.2920
11. Forlino A, Marini JC. Osteogenesis imperfecta. Lancet. 2016;387(10028):1657-1671. doi:10.1016/S0140-6736(15)00728-X
12. NM_000089.4(COL1A2):c.964G>A (p.Gly322Ser). National Center for Biotechnology Information. February 7, 2023. Accessed November 20, 2023. https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000618023.18
13. Isidor B, Lindenbaum P, Pichon O, et al. Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat Genet. 2011;43(4):306-308. doi:10.1038/ng.778
14. McDaniell R, Warthen DM, Sanchez-Lara PA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79(1):169-173. doi:10.1086/505332
15. NM_024408.4(NOTCH2):c.3467A>G (p.Asn1156Ser). National Center for Biotechnology Information. April 15, 2023. Accessed November 20, 2023. https://www.ncbi.nlm.nih.gov/clinvar/RCV000121725/
16. Di Marcello F, Di Donato G, D’Angelo DM, Breda L, Chiarelli F. Bone health in children with rheumatic disorders: focus on molecular mechanisms, diagnosis, and management. Int J Mol Sci. 2022;23(10):5725. doi:10.3390/ijms23105725
AFFILIATIONS:
1Medical student, University of South Florida Health Morsani College of Medicine, Tampa, FL
2Associate Professor, Department of Pediatrics, University of South Florida Health Morsani College of Medicine, Tampa, FLCITATION:
Fiedler CR, Soylu L. An unexpected outcome in an adolescent with juvenile ankylosing spondylitis. Consultant. 2024;64(2):e1. doi:10.25270/con.2024.01.000005
Received September 26, 2023. Accepted December 4, 2023. Published online January 23, 2024.DISCLOSURES:
The authors report no relevant financial relationships.ACKNOWLEDGEMENTS:
None.CORRESPONDENCE:
Cole R. Fiedler, 1513 S Bay Villa Place, Tampa, FL 33629 (colefiedler@usf.edu)© 2023 HMP Global. All Rights Reserved.
Any views and opinions expressed are those of the author(s) and/or participants and do not necessarily reflect the views, policy, or position of Consultant360 or HMP Global, their employees, and affiliates.