
Peer Reviewed
Seronegative Atypical Diffuse Systemic Sclerosis
Introduction. A 54-year-old man presented to a rheumatology clinic for ongoing care.
Patient history. The patient initially presented to the rheumatology clinic 9 years earlier with a 7-year history of diffuse painful skin. Upon his initial visit, the patient stated that a previous dermatologist suggested a diagnosis of systemic sclerosis (SSc), but the patient did not have this diagnosis confirmed. Now seeking confirmation, he presented to our clinic for further evaluation and management. He denied a history of exposure to vinyl chloride, toxic oil, tryptophan, bleomycin, chemical solvents, pentazocine, and tick bites. He had exposure to gadolinium during a previous magnetic resonance imaging performed about 13 years before he developed symptoms. He also noted a possible exposure to silica dust while doing construction work prior to developing symptoms; however, he could not confirm that silica was present in the construction materials.
At the current visit, he stated a history of painful leg ulcers, finger contractures with decreased range of motion, painful progressing calcinosis, worsening pain of Raynaud phenomenon (decreased blood flow to the fingers), and pain in bilateral hips and lower back. On physical examination, the patient had pink-white ulcerated sclerotic plaques on the left posterior distal shin (Figure 1A) and right medial malleolus (Figure 1B), diffuse calcified nodules, scattered telangiectasias on the face, chest, and hands, sclerodactyly, pinched nose, decreased size of oral aperture, and diffuse taut, shiny, thickened skin (Figure 2A-D).
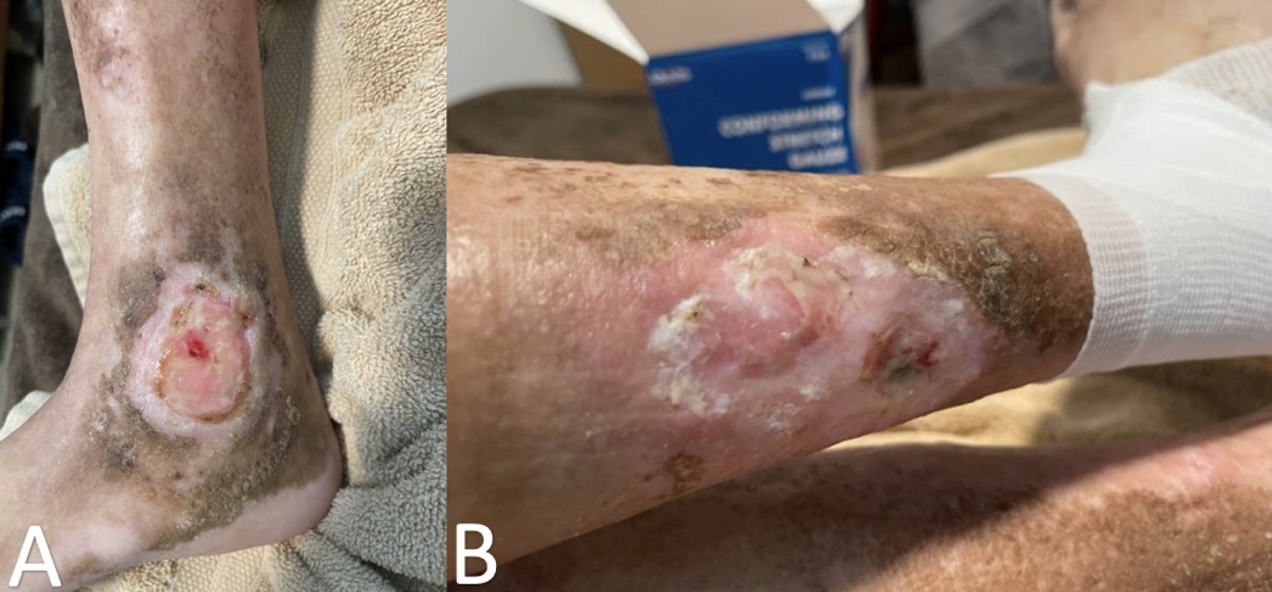
Figure 1A-B. Ulcerated plaque on left posterior distal shin is shown (Figure 1A). Additionally, an ulcerated plaque on right medial malleolus can be seen (Figure 1B).
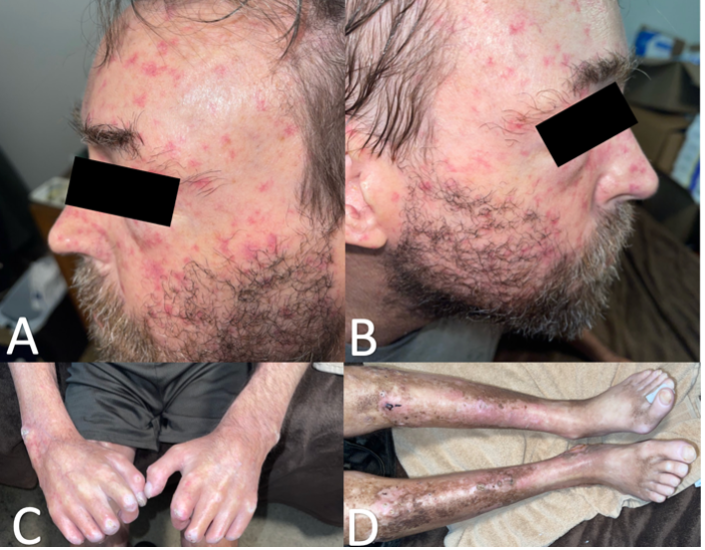
Figure 2A-D. Scattered telangiectasias across the left (Figure 2A) and right (Figure 2B) side of face and thinning of distal eyebrow hair. Sclerodactyly of right and left digits and calcified nodules on right lateral wrist, left lateral wrist, right first interphalangeal joint, right 2nd-4th proximal interphalangeal joints, and left 2nd and 3rd proximal interphalangeal joints are shown (Figure 2C). Taut, shiny, thickened skin of bilateral lower extremities with overlying hyperpigmentation and erythema are shown (Figure 2D).
A review was done of the patient’s medications. He has a history of hypertriglyceridemia, which was being treated with fenofibrate 134 mg once a day. The patient’s additional comorbidities included complications associated with SSc, particularly hypertension, gastroesophageal reflux disease (GERD), insomnia, gastrointestinal dysmotility, depression, and chronic pain. To manage these SSc-related complications, he was being treated with sildenafil 20 mg three times a day, nifedipine 60 mg twice a day, pantoprazole 40 mg once a day, clopidogrel 75 mg once a day (with re-evaluation at each visit), temazepam 15 mg at bedtime as needed, loperamide 2 mg as needed (2 mg taken after first loose stool, followed by 2 mg after each subsequent loose stool; not to exceed 16 mg per day), venlafaxine 75 mg once a day for 4 weeks (followed by re-evaluation), and oxycodone-acetaminophen 5 mg-325 mg three-to-four times a day (followed by re-evaluation at each visit).
Diagnostic testing. The patient stated during his initial visit that he had a negative autoimmune laboratory work-up in the past, so serologies were redrawn. The results showed negative results for the following: anti-centromere, anti-cyclic citrullinated peptide, anti-double stranded-DNA, anti-La/SSB, antinuclear, rheumatoid factor, anti-ribonucleoprotein, anti-RNA polymerase III, anti-Ro/SSA, anti-Scleroderma 70 (topoisomerase I), and anti-Smith.
Differential diagnoses. When this patient initially presented 9 years ago, a comprehensive work-up and diagnostic testing was done. Given this patient’s history and clinical findings and despite the negative autoimmune serologies, using the American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) criteria, which classifies patients with a score of 9 or greater as having SSc (Table 1), it was felt that the patient’s diagnosis was most consistent with SSc, due to his score of 18.
Regarding the ACR/EULAR 2013 criteria, the total score is determined by adding the maximum score in each category. Patients having a total score of 9 or greater are classified as having definite SSc. Therefore item 1, or “skin thickening of the fingers of both hands extending proximal to the metacarpophalangeal joints,” is a sufficient criterion with a score of 9. Items 2, “skin thickening of the fingers,” and 3, “fingertip lesions,” both have sub-items with different scores. The total score must only include the higher score for that sub-item (i.e., if someone with fingertip lesions has both digital tip ulcers (2 points) and fingertip pitting scars (3 points), their total score for that item is 3). Items 6, “pulmonary arterial hypertension and/or interstitial lung disease,” and 8, “SSc-related antibodies,” both have sub-items with the same scores. Therefore, for each item, there is a maximum score that can be counted towards the total score (i.e. if someone with SSc-related antibodies has anticentromere (3 points) and anti-topoisomerase I antibodies (3 points), their maximum score for that item is 3). Exclusionary criteria: These criteria are applicable to any patient considered for inclusion in a SSc study. These criteria are not applicable to patients having a SSc-like disorder that better explains their manifestations or patients with skin thickening that spares the fingers.
Table 1. The ACR/EULAR 2013 criteria for the classification of systemic sclerosis5
Item | Sub-item | Score | Patient Score |
1. Skin thickening of the fingers of both hands extending proximal to the metacarpophalangeal joints (sufficient criterion) |
| 9 | 9 |
2. Skin thickening of the fingers (only count the higher score) | Puffy fingers | 2 | 0 |
Sclerodactyly of the fingers (distal to the metacarpophalangeal joints but proximal to the proximal interphalangeal joints) | 4 | 4 | |
3. Fingertip lesions (only count the higher score) | Digital tip ulcers | 2 | 0 |
Fingertip pitting scars | 3 | 0 | |
4. Telangiectasia |
| 2 | 2 |
5. Abnormal nailfold capillaries |
| 2 | 0 |
6. Pulmonary arterial hypertension and/or interstitial lung disease (maximum score is 2) | Pulmonary arterial hypertension | 2 | 0 |
Interstitial lung disease | 2 | 0 | |
7. Raynaud phenomenon |
| 3 | 3 |
8. SSc-related antibodies (maximum score is 3) | Anticentromere | 3 | 0 |
Anti-topoisomerase I | 3 | 0 | |
Anti-RNA polymerase III | 3 | 0 |
Differential diagnoses that were considered were pseudoscleroderma conditions, or SSc mimickers (Table 2). These conditions were ruled out through the patient’s history and physical examination.
Table 2. Scleroderma-like disorders8,9
Disease | Clinical Features |
Eosinophilic fasciitis | Acute scleroderma-like skin changes with painful, symmetrical swelling and progressive induration of the extremities with peripheral eosinophilia, hypergammaglobulinemia, and elevated sedimentation rate. |
Sclerodermiform genodermatoses | Stiff, thin skin with an atrophic surface and moderate hyperkeratotic areas, lower extremity ulcers or calluses, thinning and graying of hair or baldness, nail dystrophy or loss, and wrinkling and aging of the face (bird-like face). |
Acrodermatitis chronica atrophicans | Late stage of Borrelia infection characterized by unilateral, darkly pigmented, edematous swelling of the lower extremity or forearm that progresses into an atrophic stage with cigarette paper-like skin. |
Scleredema (scleredema adultorum Buschke) | Symmetrical, erythematous, diffuse, progressive, non-pitting swelling and induration of the nape of the neck, shoulders, and upper limbs with sparing of the hands and feet. Associated with diabetes mellitus, history of upper respiratory tract infection, or blood dyscrasia. |
Scleredema diabeticorum | Bilateral thickened pebble-like skin and grouped waxy papules on fingers and hands with joint contractures in severe forms. Associated with diabetes. |
Scleromyxedema | Firm, smooth papules on limbs, trunk, and face with frequent internal organ involvement and no Raynaud’s phenomenon. |
Nephrogenic fibrosing sclerosis | Painful, pruritic, hyperpigmented skin with papules that may coalesce to form symmetrical patches and plaques with a sclerodermoid and peau d’orange appearance, commonly on the extremities and trunk, sparing the face. Associated with moderate-to-severe (acute or chronic) kidney failure, especially in patients on dialysis and/or with exposure to gadolinium-based contrast agents. |
Porphyria cutanea tarda | Indurated, waxy, yellowish plaques that resemble scleroderma and may develop dystrophic calcifications in light-exposed and covered skin, most prominent on preauricular and nuchal areas, V-shaped area of chest, hands, and face. |
Sclerodermatous graft-versus-host disease | Circumscribed firm plaques starting on the lower trunk (localized form) then spreading to the entire trunk and proximal extremities and causing a progressive restrictive lung disease (generalized form). The generalized form may be preceded by a lichenoid, erythematous, violaceous eruption. |
Treatment and management. Since initial presentation, the patient’s condition has progressively worsened. He now experiences recurrent cellulitis and infections, requiring frequent use of oral and/or IV antibiotics, which vary depending on the infection. In addition to at-home wound care and routine rheumatology follow-ups, hyperbaric oxygen chamber therapy is being considered as a possible option by the patient to reduce inflammation and alleviate ulcer and joint pain. To minimize disease progression, primary care of this patient involves symptomatic management, including the recommended continued use of current medications with re-evaluation at each visit and ongoing monitoring for organ involvement.
Outcome and follow-up. Systemic sclerosis complications remain relatively controlled compared with what is expected for a patient with this severity of cutaneous involvement. Previous imaging studies revealed cardiac, vascular, and pulmonary involvement, although he remains asymptomatic. An echocardiogram performed 1-year earlier revealed a pericardial effusion. A computed tomography (CT) angiogram of the abdomen and pelvis performed 1-year earlier revealed bulky calcifications of the superior mesenteric artery, splenic artery, abdominal aorta, bilateral renal arteries, and common iliac arteries. A CT scan of the chest performed 3-years ago demonstrated ground-glass opacities and reticulations along the periphery of bilateral lung bases. He continues to be monitored with annual pulmonary function tests, echocardiograms, and CT angiograms of the abdomen and pelvis, as well as annual labs, including a complete blood count with erythrocyte sedimentation rate, comprehensive metabolic panel, and lipid panel.
Discussion. Systemic sclerosis is an immune-mediated connective tissue disease that typically features progressive skin fibrosis. This condition is clinically classified into two main types: limited SSc (lcSSc), also known as CREST syndrome, and diffuse SSc (dcSSc). In lcSSc, skin thickening is confined to the distal limbs and face and is accompanied by calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasias. Patients typically have positive anti-centromere and antinuclear antibodies. Alternatively, dcSSc is characterized by more rapid, diffuse skin thickening typically found across the distal and proximal extremities, face, and trunk. Organ involvement is more common in dcSSc, including interstitial lung disease, pulmonary arterial hypertension, scleroderma renal crisis, and cardiac involvement. Associated antibodies are anti-topoisomerase/Scl-70, anti-RNA polymerase III, and antinuclear.1
Several environmental factors have been identified that lead to SSc or SSc-like conditions, including silica dust, vinyl chloride, tryptophan, gadolinium, bleomycin, and pentazocine. The method of how these compounds trigger these diseases is still unknown.2,3 Additionally, exposure to Borrelia Burgdorferi species, which most commonly occurs via tick bites, has been associated with chronic sclerodermiform states, most commonly plaque scleroderma.4
The ACR/EULAR defined a set of classification criteria in 2013 to assist in evaluation of patients with SSc symptoms. According to the ACR/EULAR, these criteria are not applicable to patients with finger-sparing skin thickening or who have a SSc-like disorder that better explains their symptoms. Patients with a score of 9 or greater are classified as having SSc. However, as the complexity of the disease is highly variable, these criteria are not definitive.5 Still, the ACR/EULAR recommend that SSc therapies focus on targeting the symptomatic complications and minimizing disease progression.
As demonstrated in this case, seronegative SSc is extremely rare. Antinuclear antibodies (ANA) are positive in more than 90% of patients diagnosed with SSc. However, the current SSc criteria do not include the presence of ANA. Therefore, its negativity does not exclude the diagnosis.6 There are few studies analyzing the prevalence of seronegative SSc patients. A 2013 multicenter prospective cohort study conducted by the EULAR Scleroderma Trials and Research (EUSTAR), which analyzed 5390 patient reports, found that only 7.7% of patients with SSc had a negative ANA. Of those, 0.2% of patients had neither a history of Raynaud phenomenon nor a positive ANA. Notably, within this 0.2% subset of patients, 66.7% had a phenotype consistent with dcSSC.7 ANA-negative SSc is associated with distinct characteristics, including less vasculopathy and a higher frequency of lower gastrointestinal involvement. Additionally, the disease course is often more severe and occurs at a higher proportion in men.7
Systemic sclerosis diagnosis requires a comprehensive work-up to rule out pseudosclerodermas and an evaluation for an overlap syndrome.8,9 Approximately 10% of patients with SSc have clinical features of an overlap syndrome, most commonly mixed connective tissue disease, which is characterized by skin involvement that rarely spreads proximal to the wrists, Raynaud phenomenon, positive ANA and/or anti-U1RNP antibodies, and puffy hands or sclerodactyly.
In cases where SSc-like symptoms are present without Raynaud phenomenon or a positive ANA, a paraneoplastic syndrome should be considered. Indeed, EULAR acknowledges that while there are reports of associations between SSc and various malignancies, the relationship is not well established. Additionally, SSc lesions have been linked to various malignancies, including breast, lung, hematologic, oropharyngeal, esophageal, and gastrointestinal. Specifically, dcSSc is associated with cancers of the breast, uterus, and alveolar and bronchial lung tumors, while lcSSc is associated with carcinoid and bronchoalveolar lung tumors.9 Notably, the onset of skin involvement often precedes clinical malignancy manifestations, underscoring the importance of evaluating patients presenting with SSc-like lesions for underlying malignancies.
Conclusion. The diagnosis and classification of SSc is a challenging and continuously evolving process. A comprehensive clinical evaluation, including detailed history-taking, a thorough physical examination, nail-fold capillaroscopy, serological testing, skin biopsy, and chest CT is imperative when suspecting SSc.7 Pseudosclerodermas must be ruled out, and malignancy evaluation should be conducted, especially in those with negative serology and/or no history of Raynaud’s phenomenon. However, SSc treatment remains to treat the symptoms arising from any organ complications and minimize disease progression, therefore each patient should be evaluated and treated on an individualized case-by-case basis.
- Jerjen R, Nikpour M, Krieg T, Denton CP, Saracino AM. Systemic sclerosis in adults. Part I: Clinical features and pathogenesis. J Am Acad Dermatol. 2022;87(5):937-954. doi: 10.1016/j.jaad.2021.10.065
- Rosendahl AH, Schönborn K, Krieg T. Pathophysiology of systemic sclerosis (scleroderma). Kaohsiung J Med Sci. 2022;38(3):187-195. doi: 10.1002/kjm2.12505.
- Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol. 2012;24(2):165-70. doi: 10.1097/BOR.0b013e32834ff2e8.
- Haddad V Jr, Haddad MR, Santos M, Cardoso JLC. Skin manifestations of tick bites in humans. An Bras Dermatol. 2018;93(2):251-255. doi: 10.1590/abd1806-4841.20186378.
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737-47. doi: 10.1002/art.38098.
- Cavazzana I, Vojinovic T, Airo' P, et al. Systemic sclerosis-specific antibodies: novel and classical biomarkers. Clin Rev Allergy Immunol. 2023;64(3):412-430. doi: 10.1007/s12016-022-08946-w.
- Schneeberger D, Tyndall A, Kay J, et al. Systemic sclerosis without antinuclear antibodies or Raynaud's phenomenon: a multicentre study in the prospective EULAR Scleroderma Trials and Research (EUSTAR) database. Rheumatology (Oxford). 2013;52(3):560-7. doi: 10.1093/rheumatology/kes315.
- Fabri M, Hunzelmann N. Differential diagnosis of scleroderma and pseudoscleroderma [in German]. J Dtsch Dermatol Ges. 2007;5(11):977-984. English, German. doi: 10.1111/j.1610-0387.2007.06311.x.
- Ferreli C, Gasparini G, Parodi A, Cozzani E, Rongioletti F, Atzori L. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53(3):306-336. doi: 10.1007/s12016-017-8625-4
AUTHORS:
Kyra Diehl BS1 • Oliver J. Wisco DO, FAAD, FACMS2 • Cara Barber MD3 • Daniel E. Furst MD4
AFFILIATIONS:
1Western University of Health Sciences, College of Osteopathic Medicine, Pomona, CA
2Department of Dermatology, Warren Alpert Medical School of Brown University, Providence, RI
3Silver Falls Dermatology, Salem, OR
4Division of Rheumatology, University of California, Los Angeles, Los Angeles, CA
CITATION:
Diehl K, Wisco OJ, Barber C, Furst DE. Seronegative atypical diffuse systemic sclerosis. Consultant. Published online August 30, 2024. doi:10.25270/con.2024.08.000006
Received March 14, 2024. Accepted May 14, 2024
DISCLOSURES:
The authors report no relevant financial relationships.
ACKNOWLEDGEMENTS:
None.
CORRESPONDENCE:
Kyra Diehl, BS, 6601 Avenida La Reina, La Jolla, CA 92037 (kyra.diehl@westernu.edu)