
Peer Reviewed
What Are This Woman’s Widespread Asymptomatic Nodules?
Authors:
Alexander K. C. Leung, MD; Benjamin Barankin, MD; and Amy A. M. Leung, OD
Citation:
Leung AKC, Barankin B, Leung A. What are this woman’s widespread asymptomatic nodules? Consultant. 2017;57(11):651-654.
A 54-year-old Chinese woman presented with primarily cosmetic concerns about numerous brownish spots and nodules on her body. She found the nodules socially embarrassing and wondered whether the nodules could be removed noninvasively. She recalled that the brownish spots had been present since early childhood, and that the nodules had become more apparent at age 25 years, when she had been pregnant. The brownish spots and nodules had increased in size and number with time.
Findings of a review of systems were unremarkable; there was no functional impairment. Her 24-year-old daughter also had had brownish spots at birth, and a few soft skin nodules had developed a few years ago.
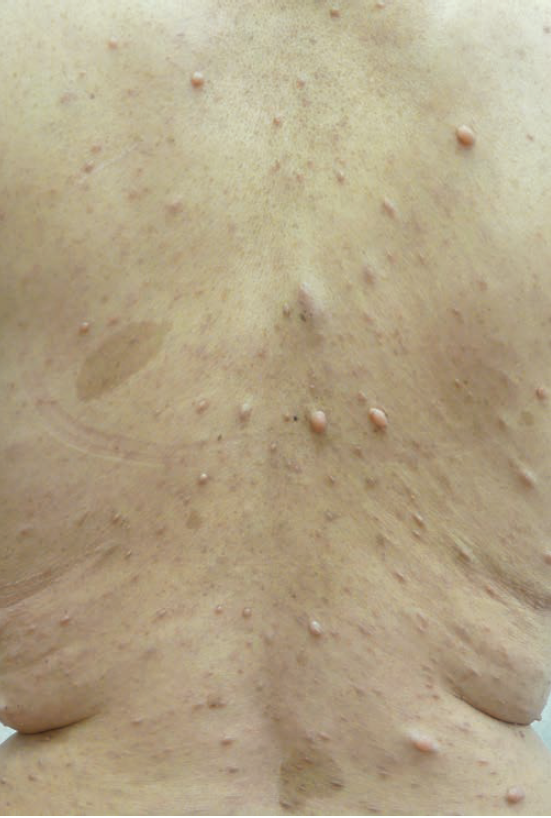
Physical examination revealed 15 café au lait spots greater than 1.5 cm in diameter; numerous soft, skin-colored nodules on the trunk and extremities; 2 large, ill-defined, soft, subcutaneous nodules, one on each side on the lower back; and bilateral axillary freckling. Slit-lamp ophthalmologic examination revealed several well-defined, brownish, dome-shaped nodules rising from the surface of the iris bilaterally. Her blood pressure was 130/85 mm Hg. No other cutaneous or systemic abnormalities were present.
Answer on next page
Answer: Neurofibromatosis Type 1
Based on the clinical findings of 15 café au lait spots greater than 1.5 cm in diameter, numerous cutaneous neurofibromas, 2 plexiform neurofibromas, bilateral axillary freckling, bilateral Lisch nodules, and a positive family history, a diagnosis of neurofibromatosis type 1 (NF1) was made. Several neurofibromas were excised for cosmetic purposes. The patient was advised to come back annually to have the plexiform neurofibromas and her blood pressure monitored.
Overview. NF1, previously known as von Recklinghausen disease, is characterized by 6 or more café au lait spots, intertriginous freckling, 2 or more iris hamartomas (Lisch nodules), 2 or more neurofibromas of any type or 1 plexiform neurofibroma, a distinct osseous lesion, and a positive history of NF1 in a first-degree relative.1,2 NF1 was first recognized as a distinct disease entity by Friedrich Daniel von Recklinghausen in 1882.3 The condition is known as segmental NF1 if its clinical features are confined to a single area of the body.4,5
Epidemiology. NF1 is the most prevalent neurocutaneous syndrome and accounts for about 90% of all cases of neurofibromatosis.2 NF1 occurs in 1 of every 2500 to 3500 live births.6,7 Most patients present before 8 years of age.2 Late-onset NF1 has rarely been reported.8 There is no gender or racial predilection.9
Pathogenesis. NF1 is inherited in an autosomal dominant fashion with complete penetrance.7,10 The neurofibromin 1 gene (NF1) has been mapped to 17q11.2.5,11 Approximately 50% of cases are sporadic and attributable to a new mutation.4,12 NF1 directs the synthesis of neurofibromin, a Ras GTPase-activating protein, which has a role in tumor suppression. Neurofibromin is expressed mainly in Schwann cells, astrocytes, oligodendrocytes, leukocytes, and the adrenal medulla.6 Inactivation of the gene, either through allelic loss or mutation, results in reduced neurofibromin production or function, which leads to uncontrolled cell growth or tumor formation.2 Neurofibromin is also involved in the differentiation of the neuronal neurotransmitter phenotype and in synapse formation.2
CLINICAL MANIFESTATIONS
Café au lait spots. These ovoid, uniformly brown macules are found in almost all patients with NF1.4,13 They are frequently present at birth and increase in size, number, and pigmentation with age. There is a predilection for the trunk and extremities.13 The scalp, face, palms, and soles are usually spared. The number and location of café au lait spots have no relationship to the number and location of future neurofibromas.13 However, a large hyperpigmented lesion with an irregular border that is present at birth might herald an underlying plexiform neurofibroma.13
Neurofibromas. These benign peripheral nerve-sheath tumors are composed of an extracellular matrix and a heterogeneous mixture of Schwann cells and fibroblasts.4,6 Cutaneous neurofibromas often appear during puberty or pregnancy, increase in size and number with age, and are found in almost all adults with NF1.14 These painless, soft or elastic, flesh-colored, nodular or pedunculated lesions often invaginate into the skin and exhibit the buttonhole sign when gentle digital pressure is applied to the surface.4 In contrast, subcutaneous neurofibromas, which are usually firm and painful, are deeply seated in the dermis and, therefore, less circumscribed. Plexiform neurofibromas are often present at birth and result from diffuse thickening of nerve trunks and feel like “a bag of worms.”6 They are found in 30% to 50% of individuals with NF1.6 Plexiform neurofibromas may be locally invasive and associated with bone erosion and pain, spinal cord compression, and overgrowth of an extremity.
Intertriginous freckling. Freckles, usually in clusters, develop in the axillae, inguinal areas, or submammary regions by late childhood in approximately 80% of individuals with NF1.6 Intertriginous freckling is not related to sun exposure and is pathognomonic of NF1 (the Crowe sign).2
Optic gliomas and other intracranial tumors. Optic gliomas occur in approximately 15% of children younger than 6 years with NF1.5,9,11 These tumors are low-grade pilocytic astrocytomas; they tend to grow slowly and are usually asymptomatic.10,11,15 In approximately 12% of affected individuals, visual disturbance, proptosis, or precocious puberty develops; the most common presenting symptom is asymmetric noncorrectable vision loss.10 With a unilateral lesion, there may be an ipsilateral relative afferent pupillary defect. Other intracranial tumors include astrocytomas, brainstem gliomas, cerebellar gliomas, ependymomas, meningiomas, and neurilemmomas.5,6,15
Lisch nodules. Lisch nodules are melanocytic hamartomas of the iris and present as yellow-brown, dome-shaped elevations on the iris surface. They are best identified by way of slit-lamp examination or dermoscopy.16 Lisch nodules do not cause visual disturbance. They are present in more than 90% of adults with NF1 and are usually bilateral.5,6,16
Musculoskeletal abnormalities. Osseous lesions include sphenoid wing dysplasia, scoliosis, cortical thinning of long bones, dysplasia of long bones, and pseudarthrosis.6,11 Other skeletal abnormalities include macrocephaly, megadactyly, pectus excavatum, and osteopenia/osteoporosis.14 Short stature is common.11
Neurofibromin plays an important role in muscle metabolism and muscle development.17,18 Reduced muscle size, muscle weakness, hypotonia, and poor fine/gross motor coordination are increasingly recognized as common manifestations of NF1.17,18
Learning disabilities. Approximately 50% to 75% of patients with NF1 have learning disabilities and cognitive deficits.14 Abnormal visual-spatial performance, social competence, receptive and expressive language problems, reading and writing difficulties, attention-deficit/hyperactivity disorder, incoordination, and clumsiness are common in individuals with NF1.2,9,14 Intellectual disability is only slightly more common than it is in the general population. In 60% to 70% of patients with NF1, magnetic resonance imaging demonstrates unidentifiable bright objects, frequently in the basal ganglia, brainstem, subcortical white matter, and cerebellum.2 Presumably, they represent increased fluid within the myelin, associated with dysplastic glial proliferation.5 The lesions do not exert any mass effect and often resolve over time.
Seizures. The prevalence of seizures in persons with NF1 is approximately 4% to 13%.6,9 Complex partial seizures are the most common form.
Cardiovascular abnormalities. Congenital heart disease (eg, valvular pulmonary artery stenosis, hypertrophic cardiomyopathy) and vasculopathy (eg, arteriovenous malformation, moyamoya disease) occur with increased frequency in NF1 patients.6,14 Approximately 6% of persons with NF1 have hypertension, which may be secondary to renal artery stenosis, coarctation of the aorta, or pheochromocytoma.6
Endocrine abnormalities. Precocious puberty, diencephalic syndrome, growth hormone deficiency, growth hormone hypersecretion, and gynecomastia are more common than expected in patients with NF1.7
Miscellaneous. Juvenile xanthogranuloma, nevus anemicus, glomus tumor, insomnia, and headaches occur with increased frequency in persons with NF1.5,10,14
DIAGNOSIS
NF1 is diagnosed clinically based on National Institutes of Health (NIH) criteria (Table).1 To make the diagnosis of NF1, 2 or more of the diagnostic criteria must be present.1 The diagnosis of NF1 can be made confidently using the NIH diagnostic criteria by 8 years of age in most children.19 Apart from identification of the NF1 gene, no laboratory test confirms the diagnosis. DNA analysis of the gene is neither widely available nor clinically helpful in most situations. Gene testing may be helpful in prenatal diagnosis.

Differential Diagnosis
Café au lait spots, the most important hallmark of NF1, are also found in neurofibromatosis type 2, McCune-Albright syndrome, tuberous sclerosis complex, multiple mucosal neuroma syndrome, Fanconi anemia, ataxia telangiectasia, Bloom syndrome, Legius syndrome, Noonan syndrome, Proteus syndrome, Klippel-Trénaunay-Weber syndrome, Watson syndrome, constitutional mismatch repair deficiency syndrome, and LEOPARD (multiple lentigines) syndrome.4,12,14 The distinctive features of each condition allow a fairly straightforward differentiation from NF1.
Complications
Café au lait spots and neurofibromas can be disfiguring and socially embarrassing for patients if the lesions affect a visible area. The resultant anxiety, distress, depression, and social isolation have a significant adverse effect on the quality of life.18,20 Affected patients have lower self-esteem compared with the general population.21
Dumbbell-shaped neurofibromas can develop in the spinal cord and cause compression syndromes.5 Patients with NF1 are at increased risk for malignancies, such as myelomonocytic leukemia, pheochromocytoma, rhabdomyosarcoma, carcinoid tumor, Wilms tumor, neuroblastoma, breast cancer, gastrointestinal stromal tumors, and malignant peripheral nerve-sheath tumor.5,12,15,20 The lifetime risk of plexiform neurofibromas transforming into malignant peripheral nerve-sheath tumors ranges from 5% to 13%.5,11
Prognosis
NF1 is a progressive disorder with clinical features accumulating over time. The prognosis depends on the severity of the disease and the systemic involvement. The life expectancy of patients with NF1 is reduced by 8 to 15 years relative to the general population, mainly as a result of malignancy.6,11
Management
The mainstay of management is anticipatory guidance.6 A multidisciplinary approach is essential and may require the expertise of a dermatologist, ophthalmologist, neurologist, orthopedic surgeon, plastic surgeon, geneticist, and pediatrician/internist. Although no cure for NF1 currently exists, laser or traditional surgery can reduce the appearance of cutaneous neurofibromas and lasers can in some cases fade café au lait macules and patches.
All first-degree relatives of an affected patient should be examined for cutaneous manifestations and should undergo a slit-lamp ophthalmologic examination to determine whether the case is sporadic or familial. Patients with a family history of NF1 should be informed that the recurrence rate is 50%. For sporadic cases, the risk of another affected child is low and is based on the rare occurrence of germinal mosaicism.
Alexander K. C. Leung, MD, is clinical professor of pediatrics at the University of Calgary and a pediatric consultant at the Alberta Children’s Hospital in Calgary, Alberta, Canada.
Benjamin Barankin, MD, is a dermatologist and the medical director and founder of the Toronto Dermatology Centre in Toronto, Ontario, Canada.
Amy A. M. Leung, OD, is a medical student at the University of Alberta in Edmonton, Alberta, Canada.
References:
- Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278(1):51-57.
- Leung AKC, Robson WLM. A young girl with café au lait spots. Consultant Pediatricians. 2006;5(4):229-232.
- von Recklinghausen F. Ueber die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen. Berlin, Germany: August Hirschwald, 1882.
- Hernández-Martín A, Duat-Rodríguez A. An update on neurofibromatosis type 1: not just café-au-lait spots, freckling, and neurofibromas. An update. Part I. Dermatological clinical criteria diagnostic of the disease. Actas Dermosifiliogr. 2016;107(6):454-464.
- Korf BR. Neurofibromatosis type 1 (NF1): pathogenesis, clinical features, and diagnosis. UpToDate. https://www.uptodate.com/contents/neurofibromatosis-type-1-nf1-pathogenesis-clinical-features-and-diagnosis. Updated September 27, 2017. Accessed October 17, 2017.
- Anderson JL, Gutmann DH. Neurofibromatosis type 1. Handb Clin Neurol. 2015;132:75-86.
- Bizzarri C, Bottaro G. Endocrine implications of neurofibromatosis 1 in childhood. Horm Res Paediatr. 2015;83(4):232-241.
- Uchiyama M, Sakai N, Maeda T, Tsuboi R, Mitsuhashi Y. Late-onset neurofibromatosis revealing NF1 mutation. J Dermatol. 2015;42(1):104-105.
- Ferner RE, Gutmann DH. Neurofibromatosis type 1 (NF1): diagnosis and management. Handb Clin Neurol. 2013;115:939-955.
- Hernández-Martín A, Duat-Rodríguez A. An update on neurofibromatosis type 1: not just café-au-lait spots and freckling. Part II. Other skin manifestations characteristic of NF1. NF1 and cancer. Actas Dermosifiliogr. 2016;107(6):465-473.
- Kresak JL, Walsh M. Neurofibromatosis: a review of NF1, NF2, and schwannomatosis. J Pediatr Genet. 2016;5(2):98-104.
- Ben-Shachar S, Dubov T, Toledano-Alhadef H, et al. Predicting neurofibromatosis type 1 risk among children with isolated café-au-lait macules. J Am Acad Dermatol. 2017;76(6):1077-1083.e3.
- Leung AKC. Café au lait macules. In: Leung AKC, ed. Common Problems in Ambulatory Pediatrics: Specific Clinical Problems. Vol 2. New York, NY: Nova Science Publishers; 2011:223-227.
- Friedman JM. Neurofibromatosis 1. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2014. https://www.ncbi.nlm.nih.gov/books/NBK1109/. Accessed October 17, 2017.
- Varan A, Şen H, Aydın B, Yalçın B, Kutluk T, Akyüz C. Neurofibromatosis type 1 and malignancy in childhood. Clin Genet. 2016;89(3):341-345.
- Gómez Moyano E, Martínez Pilar L, Rodriguez Calvo de Mora M, et al. Using dermoscopy to assess diagnostic criteria of neurofibromatosis. J Am Acad Dermatol. 2015;73(1):e17-e18.
- Cornett KMD, North KN, Rose KJ, Burns JM. Muscle weakness in children with neurofibromatosis type 1. Dev Med Child Neurol. 2015;57(8):733-736.
- Summers MA, Quinlan KG, Payne JM, Little DG, North KN, Schindeler A. Skeletal muscle and motor deficits in neurofibromatosis type 1. J Musculoskelet Neuronal Interact. 2015;15(2):161-170.
- DeBella K, Szudek J, Friedman JM. Use of the National Institutes of Health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000;105(3 pt 1):608-614.
- Ferner RE, Thomas M, Mercer G, et al. Evaluation of quality of life in adults with neurofibromatosis 1 (NF1) using the Impact of NF1 on Quality of Life (INF1-QOL) questionnaire. Health Qual Life Outcomes. 2017;15(1):34.
- Rosnau K, Hashmi SS, Northrup H, Slopis J, Noblin S, Ashfaq M. Knowledge and self-esteem of individuals with neurofibromatosis type 1 (NF1). J Genet Couns. 2017;26(3):620-627.
- Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. 2001;68(5):1110-1118.