
Evans Syndrome
A 24-year-old woman with a history of menorrhagia presented to the emergency department with worsening shortness of breath, fatigue, and dizziness that had begun 3 days prior with the onset of her menstrual cycle.
Physical examination. At presentation, the patient was tachycardic, with 100% oxygen saturation on room air. She was actively menstruating, and petechiae were noted on her oral mucosa, left breast, anterior neck, and lower extremities.
Diagnostic tests. Initial laboratory test findings included a hemoglobin level of 4.6 g/dL (reference range, 14.0-17.5 g/dL), a hematocrit of 16.3% (reference range, 41%-50%), a platelet count of 1 × 103/µL (reference range, 150-350 × 103/µL), a prolonged bleeding time, and a D-dimer test result that was positive for abnormal clot formation.
The patient received 1 U of blood and platelets before experiencing a hemolytic transfusion reaction. The presence of acute dyspnea prompted computed tomography angiography (CTA) of the chest, the results of which demonstrated diffuse ground-glass opacities and nodularity (Figure).
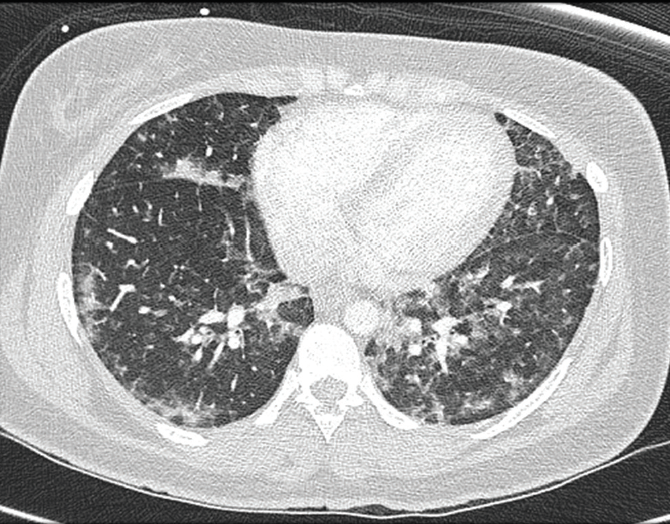
Figure. CTA of the patient’s chest demonstrated diffuse ground-glass opacities and nodularity.
The transfusion reaction in conjunction with the unusual appearance of the lungs on CTA warranted testing for autoimmune and lymphoproliferative disorders as potential causes of her symptoms.
Further testing revealed a positive direct antiglobulin test result, a positive warm reactive autoimmune antibody result, an elevated lactate dehydrogenase level, a low haptoglobin level, and an elevated reticulocyte count, all of which indicated autoimmune hemolytic anemia (AIHA) and immune/idiopathic thrombocytopenic purpura (ITP) as likely diagnoses.1
Peripheral blood smear test results were consistent with iron-deficiency anemia and severe thrombocytopenia. Flow cytometry test results of a bone marrow biopsy were unremarkable. The anemia was attributed to AIHA and iron deficiency secondary to menorrhagia. ITP was deemed to be the cause of her thrombocytopenia. The simultaneous occurrence of these 2 conditions is characteristic of Evans syndrome.1-4
Subsequent autoimmune testing revealed a positive antinuclear antibody test result, elevated single-stranded DNA antibody titers, and indeterminate double-stranded DNA antibody titers, all of which are findings suggestive of systemic lupus erythematosus (SLE), a condition frequently associated with Evans syndrome.2,4,5
Treatment. Given the severity of the patient’s anemia and thrombocytopenia, intravenous methylprednisolone and iron were initiated, which offered a slight improvement in her hemoglobin level, platelet count, and symptoms.
On day 4 of hospitalization, she was started on rituximab. In the following 2 days, she had near resolution of symptoms, and laboratory test results showed marked improvement. During that time, her hemoglobin level increased from 5.9 g/dL to 6.8 g/dL, and her platelet count increased from 8 × 103/µL to 34 × 103/µL. Upon discharge, she was transitioned to an oral corticosteroid taper, and she was followed up by a hematologist-oncologist as an outpatient to complete a 4-dose regimen of rituximab.
Discussion. Generalized fatigue and dyspnea are commonly encountered symptoms across medical specialties. Evaluation of these symptoms often may reveal anemia and thrombocytopenia. A methodical approach to determining their underlying etiology is important so as to identify less common causes such as Evans syndrome.
Evans syndrome is a rare disorder characterized by AIHA and ITP, occurring either concurrently or sequentially.1-4 While the exact occurrence rate is unknown, Evans syndrome is estimated to occur in only 0.8% to 3.7% of persons with either AHIA or ITP, and it can occur in all age groups, both sexes, and all ethnic groups.4
Although its etiology remains unclear, Evans syndrome is often associated with autoimmune disorders, immune deficiencies, or immunoproliferative disorders.2,5 In young adults, SLE is the primary condition associated with Evans syndrome. The severity of the disease varies greatly, but potential sequelae include hypoxemia, hemorrhage, and death.
Classically, the disease is chronic and relapsing despite treatment, and individual responses to each therapy vary widely.2,4 Primary treatment options include corticosteroids, intravenous immunoglobulin (IVIG), and blood or platelet transfusions. Second-line modalities include splenectomy, cytotoxic drugs, and immunosuppressive therapies and are reserved for cases that are refractory to first-line approaches.1,4
Rituximab is a chimeric monoclonal antibody to CD20 antigens on B lymphocytes. It is among the second-line agents, and it is gaining popularity for the treatment of autoimmune disorders including SLE, AIHA, and ITP.1,2,3,6
While several retrospective analyses and case reports have demonstrated that rituximab is a possible treatment option for AIHA, SLE, and Evans syndrome, no randomized controlled trials on the treatment of Evans syndrome have been performed, and structured treatment guidelines are lacking. Thus, corticosteroids and IVIG remain first-line agents for the treatment of Evans syndrome, and rituximab is reserved for cases refractory to the primary therapies.1-4
Evans syndrome should be among the differential diagnoses for any patient with severe anemia and thrombocytopenia, particularly with a history of an autoimmune, immunoproliferative, or immunodeficiency disorder, or if the anemia and thrombocytopenia have no obvious cause.
Outcome of the case. At her 3-month follow-up visit, the patient was asymptomatic, with a hemoglobin of 12.8 g/dL and a platelet count of 297 × 103/µL. She was referred to a rheumatologist for management of her SLE.
Hers is one of few reported cases in which Evans syndrome was treated with rituximab, an alternative to standard treatment regimens, prior to treatment failure or relapse. The improvement in her laboratory test results and symptomatology suggests that rituximab could be a viable treatment option for persons with Evans syndrome, without finite limitation to relapsing and refractory cases.
References:
- Schrier SL. Warm autoimmune hemolytic anemia: treatment. UpToDate. http://www.uptodate.com/contents/warm-autoimmune-hemolytic-anemia-treatment. Updated March 24, 2016. Accessed February 28, 2017.
- Costallat GL, Appenzeller S, Costallat LTL. Evans syndrome and systemic lupus erythematosus: clinical presentation and outcome. Joint Bone Spine. 2012;79(4):362-364.
- Galor A, O’Brien T. Rituximab treatment for relapsed autoimmune hemolytic anemia in Evans syndrome. Int J Hematol. 2003;78(4):335-336.
- Norton A, Roberts I. Management of Evans syndrome. Br J Haematol. 2006;132(2):125-137.
- Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114(15):3167-3172.
- Barcellini W, Zanella A. Rituximab therapy for autoimmune haematological diseases. Eur J Intern Med. 2011;22(3):220-229.