
Peer Reviewed
Panayiotopoulos Syndrome in a Pediatric Patient
AUTHORS:
Maria Camila Holguin, MD1 • Gabriela Castresana, MD2 • Carlos A. Arango, MD3
AFFILIATIONS:
1Neurology Department, Universidad CES, Medellin, Colombia
2Emergency Department, Pablo Tobon Uribe, Medellin, Colombia
3Pediatrics Department, University of Florida, Jacksonville, Florida
CITATION:
Holguin MC, Castresana G, Arango CA. Panayiotopoulos syndrome in a pediatric patient. Consultant. 2022;62(2):e8-e11. doi:10.25270/con.2021.03.00015
Received October 24, 2020. Accepted December 30, 2020. Published online March 23, 2021.
DISCLOSURES:
The authors report no relevant financial relationships.
CORRESPONDENCE:
Carlos A. Arango, MD, University of Florida, Department of Pediatrics, Baymeadows Pediatrics, 8399 Bayberry Road, Jacksonville, FL 32256 (carlos.arango@jax.ufl.edu)
A 9-year-old boy presented to the emergency department with a seizure-like episode. There were no abnormalities noted on physical examination, with an intact neurologic examination. No focal defects were found.
History. His mother reported that the patient had a regular day and normal routine and went to bed around 9:30 pm. At 10:00 pm, the patient’s brother reported the patient was vomiting. When the parents had gotten to the bedroom, the patient’s mother had taken the boy in her arms, and he had a staring episode for about 1 minute. She described the boy as being unresponsive and staring straight ahead; he also had urinated on himself during the episode. The mother reported that the patient did not appear to be experiencing fluttering, nystagmus, or eyes rolling to the back of his head, and the parents did not observe any tongue-biting or abnormal movements of his body.
On 2 nonconsecutive nights during the previous week, the patient’s parents reported that the boy had 2 episodes of nonbloody, nonbilious emesis before he went to sleep; these episodes did not present with gastrointestinal symptoms, fever, or any other concerns, the parents reported. Initially, the parents believed that food poisoning or gastritis was a possible cause of his symptoms. They also reported no history of travel, family history of neurological problems or epilepsy, or significant medical history. The boy was not taking any daily medications.
Diagnostic testing. Initial laboratory tests were conducted, results of which showed normal levels of lactic acid dehydrogenase, uric acid, and electrolytes; a normal complete blood cell count; and normal coagulation. No masses were noted on a head computed tomography (CT) scan without contrast, but a borderline low-lying cerebellar tonsil was noted. A brain magnetic resonance imaging (MRI) scan was conducted, with findings significant for a left cerebellar tonsil extending 5.2 mm below the foramen magnum and the right cerebellar tonsil extending to the level of the foramen. No surgical intervention was recommended after a neurosurgery consult.
An electroencephalogram (EEG) was conducted, with findings significant for bisynchronous posterior quadrant spikes, right hemispheric spikes, and bisynchronous with left posterior quadrant spikes. Initially, no antiepileptic medication was given.
Panayiotopoulos syndrome (PS) was diagnosed after a clinical history was taken, EEG findings were evaluated, and neuroradiological evaluation was performed. No clinical seizures were observed during admission.
Discussion. Epilepsy syndromes are a group of conditions that are different in their manifestations, diagnostic criteria, and especially in their outcomes and prognosis.1 These syndromes affect about 4% of children.2 Adults and children with epilepsy have a higher risk of mortality compared with healthy individuals. Sudden unexpected death in epilepsy (SUDEP) is a frequent epilepsy-related cause of death, although it is not as frequent in children as it is in adults.3
PS is a common, prevalent, and benign type of childhood autonomic epilepsy. It is defined as a benign age-related focal seizure disorder occurring in early and mid-childhood, especially between ages 3 and 6 years; most patients have their first seizure around age 5 years. This syndrome affects girls and boys almost equally.4
Symptoms are predominantly autonomic in nature, and electrodiagnostic studies show shifting and/or multiple foci, frequently with occipital predominance.4,5 The clinical manifestations of PS are unusual for epileptic seizures, and it is frequently mistaken with other nonepileptic diseases, such as syncope, migraine, motion sickness, and gastroenteritis, among others.6,7 It is expected that 2 to 3 out of 1000 children will be affected; these numbers could be higher if atypical cases are included.8 PS represents approximately 13% of the children with nonfebrile seizures, usually between age 3 and 6 years, and 6% of children between age 1 and 15 years.8
PS is characterized by infrequent focal seizures that are accompanied by autonomic symptoms such as emetic episodes and behavioral changes.9 Typically, this syndrome presents as an episode in which the child is conscious and reports discomfort and nausea, looks pale, and can vomit, accompanied by deviation of the head and eyes.10,11 In some cases, the child becomes unconscious, and the convulsion may progress to generalized tonic-clonic seizures.11 Two-thirds of cases occur during nocturnal sleep or during brief daytime naps, and the child wakes up with similar reports and symptoms.2,10,12 A main characteristic is that even after the most severe symptoms, the child becomes completely normal after a brief postictal sleep; this is something that helps make the diagnosis and is reassuring for the parents.13
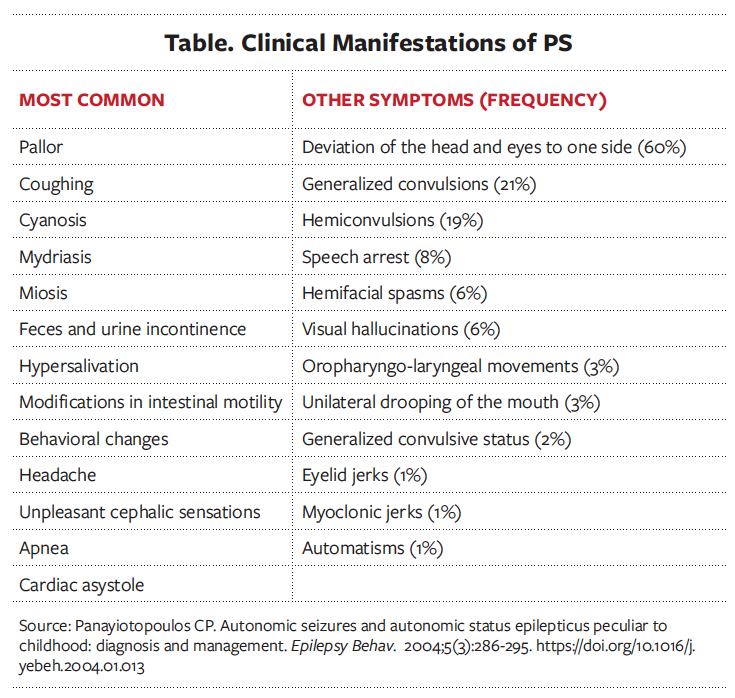
The clinical characteristics of PS are the presence of autonomic symptoms and autonomic status epilepticus (autonomic seizures that last more than 30 minutes).10,14 At the beginning of the seizure, cephalic auras may be present, as well as behavioral abnormalities such as restlessness, agitation, aggression, terror, or quietness, but the latter occurs only in 10% of children.10,14 The child may become unresponsive and confused, and the head and eyes gaze and deviate to one side.10 Headache and autonomic symptoms typically occur during the seizure.10 The most common symptom is the emetic triad that consists of nausea, retching, and vomiting.7,10 Respiratory events and cardiac arrhythmias are frequent, but cardiorespiratory arrest has been reported in very few patients.14 A list of the main symptoms of PS is shown in the Table.10
An important feature to note is that at least one-fifth of the children with PS experience ictal syncope, given by unresponsiveness and flaccidness before a seizure or even without the occurrence of one.10 In some cases, this type of syncope may be the only clinical event without other manifestations recognizable as seizures, which is why it is important to learn how to recognize it.10
The duration of the seizures varies from 1 to 60 minutes, with a mean of 9 minutes. Almost half (44%) of seizures are prolonged and can last for hours as autonomic status epilepticus.10,12 In some cases, seizures evolve from autonomic symptoms to unilateral clonic seizures associated with Jacksonian marching, or with bilateral tonic-clonic seizures. Progression to convulsive status epilepticus is rare.8,14 The duration and clinical presentation of the seizures may vary, even in the same patient, and autonomic manifestations are not always obvious.8,14 Despite the length and severity of the seizure, the child returns to normal after a few hours of sleep.8
Atypical features may be present, such as prolonged attacks for days, persistence of seizures after age 15 years, long-lasting episodes of emesis and headache, and occurrence of typical and atypical absences, among other features.15 These manifestations make the diagnosis difficult and can lead to misdiagnosis because of their resemblance to other disorders, such as cyclic vomiting syndrome, migraine, and other forms of epilepsies.15
This syndrome has an excellent prognosis. Unlike with other epilepsy syndromes, no clinical evidence has shown that lengthy seizures and status epilepticus may cause any brain damage, and they do not have any adverse prognostic significance.8,9,12 Exceptionally, atypical evolution to other epileptic syndromes and conditions of uncertain outcomes may occur.12 One-third of patients have only 1 seizure, and half of patients present with 2 to 5 seizures.9 Usually, spontaneous remission occurs within 2 years from onset, and the risk of developing epilepsy in adulthood for children with PS is the same as in the general population.9
An atypical form of PS occurs in 5% to 10% of patients; this group presents with recurrent autonomic attacks and are generally refractory to conventional antiepileptic drug therapy.9,16 Studies have shown that most of these cases have a higher incidence of mild neurobehavioral abnormalities before the onset of epilepsy, which may correlate with the refractoriness of seizures.17 It is important to detect this type of PS, since it contradicts the supposed benign nature of PS and has been described in other benign epilepsy syndromes.18
It is easy to misdiagnose and confuse PS with other common conditions.19 Early recognition of this syndrome provides reassurance to families who may be alarmed or scared, because it is a common condition in the group of benign focal epilepsies of childhood but has a high rate of misdiagnosis due to the unusual clinical manifestations.20
The diagnosis of PS is based on clinical aspects and EEG findings.20 Imaging studies, such as MRI or CT, do not show significant findings, and they are not a cornerstone in the diagnosis itself, but these neuroimaging studies can be useful for differential diagnosis.6
EEG should be performed after a first nonfebrile seizure; this study is confirmatory. Typical EEG findings in patients with PS include focal or multifocal spikes that can appear or increase during sleep. Occipital spikes predominate, but epileptiform activity may be recorded from any area and may not infrequently arise from centrotemporal regions. Occipital topography does not occur in one-third of the patients, and these kinds of spikes are not a prerequisite for the diagnosis.21 Repetitive multifocal spike-wave complexes occur in 19% of the patients, and this feature may be characteristic in this syndrome.10 Clinical manifestations are irrespective of EEG locations, and there is a variability from normal to abnormal findings even in the same patient at different times. Background EEG results are usually normal, and ictal EEG results are consistent in rhythmic θ or Δ activity, frequently intermixed with small spikes. The onset commonly is unilateral and posterior, but sometimes it is not well localized.21 It is important to acknowledge that clinical manifestations are the same regardless of the topography of spikes in the EEG scan, and occipital spikes can be seen in children with or without seizures.20
PS poses a challenge for clinicians, since it can be confused with many nonepileptic conditions, such as atypical migraine, gastroenteritis, or syncope. Prolonged and severe attacks may simulate encephalitis.13
The pathophysiology of PS is still being studied. It is suspected that this syndrome has a genetic origin, but the exact genes involved have not been identified thus far.6 A mutation in the SCN1A gene has been reported in several patients with PS. It is suspected to be involved in regulating the severity of the syndrome, and it may have a relation with fever as a trigger.22 A correlation between PS and febrile seizures has also been reported.6,14 To date, there is not yet a strong recommendation for genetic testing, because no specific related gene has been identified. It has been hypothesized that PS has a genetic origin, but more studies are needed to support this hypothesis.19
The most accepted theory for the pathophysiology of PS is that the autonomic symptoms come from an early activation of areas of the deep limbic structures that have a low threshold and are connected to the autonomic system, and then spread to other areas of the cerebral cortex.6 The fact that children with PS have such different EEG findings but show the same clinical symptoms indicates that there is a diffuse cortical hyperexcitability that is maturation related.23 The central autonomic network (CAN) is an extended circuit that controls neuroendocrine, visceromotor, behavioral, and pain responses. According to this theory, in PS there is a hyperactivation of the autonomic nervous system through the CAN, and the threshold of activation of autonomic symptomatology is lower than that in the motor or sensorial areas, which explains why an epileptic discharge activates the autonomic system, without the recruitment of neighboring cortical areas that control motor or sensory ictal manifestations.6 Clinical findings suggest that there is preferential spread to autonomic centers, including emetic centers or their cortical projections.4
Treatment and management. Because of the good prognosis of this syndrome, treatment of PS has not been well studied, and anticonvulsive treatment is usually not recommended for patients with a first seizure.16,17
Treatment with antiepileptic drugs such as valproic acid, carbamazepine, or clobazam has been recommended after a second or third seizure, depending on the length of the seizures, the association of neurobehavioral disorders, and whether the parents/guardians agree to the medication once they are informed of adverse effects related to chronic use.16 Approximately 10% to 20% of patients may present with recurring seizures or have autonomic status epilepticus for several days or with severe autonomic manifestations and may benefit from treatment with antiepileptic drugs.23 Most patients are prescribed valproic acid and carbamazepine, but recently levetiracetam has been used with excellent response achieving seizure control.23 Levetiracetam has shown the fewest adverse effects and takes less time to achieve therapeutic dose.23
For patients with prolonged seizures, it is recommended to have rescue medication at home so that parents/guardians can manage their child’s bouts of autonomic status epilepticus, such as a rectal diazepam suppository or solution.17 Other options include intramuscular, intranasal, and buccal midazolam, but diazepam is absorbed more quickly and is safer.8,24,25 Treatment modalities should be individualized to each patient.
The period of treatment should be 2 to 3 years after the last seizure. Thereafter, antiepileptic drugs are discontinued without waiting for the disappearance of epileptic EEG activity.16
Conclusion. PS is a relatively common epilepsy syndrome in childhood with a good prognosis and spontaneous remission in most cases. It is important to know about this syndrome and its manifestations because it is commonly misdiagnosed and mistaken for other common conditions. It is also important to keep in mind that making a diagnosis, giving the appropriate treatment (if needed), and counseling the patient and his or her family can improve the patient’s quality of life and allay parents’ concerns. A diagnosis is made based on clinical aspects and neurophysiologic studies. Treatment with antiepileptic drugs is not usually required, and symptoms are usually mild.
Patient outcome. The first dose of an antiepileptic drug was given to our patient in the emergency department, and then he was discharged home. He has been seizure free since initiating antiepileptic drug therapy.
References
- Wheless JW, Clarke DF, Arzimanoglou A, Carpenter D. Treatment of pediatric epilepsy: European expert opinion, 2007. Epileptic Disord. 2007;9(4):353-412. https://doi.org/10.1684/epd.2007.0144
- Koutroumanidis M. Panayiotopoulos syndrome. BMJ. 2002;324(7348):1228-1229. https://doi.org/10.1136/bmj.324.7348.1228
- Morse AM, Kothare SV. Pediatric sudden unexpected death in epilepsy. Pediatr Neurol. 2016;57:7-16. https://doi.org/10.1016/j.pediatrneurol.2016.01.004
- Ferrie C, Caraballo R, Covanis A, et al. Panayiotopoulos syndrome: a consensus view. Dev Med Child Neurol. 2006;48(03):236-240. https://doi.org/10.1017/S0012162206000508
- Saito N, Kanazawa O, Tohyama J, et al. Brain maturation-related spike localization in Panayiotopoulos syndrome: magnetoencephalographic study. Pediatr Neurol. 2008;38(2):104-110. https://doi.org/10.1016/j.pediatrneurol.2007.08.020
- Graziosi A, Pellegrino N, Di Stefano V, Raucci U, Luchetti A, Parisi P. Misdiagnosis and pitfalls in Panayiotopoulos syndrome. Epilepsy Behav. 2019;98:124-128. https://doi.org/10.1016/j.yebeh.2019.07.016
- Narayanankutty SM, Parvathy VK. Benign occipital epilepsy of childhood: Panayiotopoulos syndrome in a 3-year-old child. Int J Med Res Health Sci. 2014;3(4):1039-1043. https://doi.org/10.5958/2319-5886.2014.00049.6
- Covanis A. Panayiotopoulos syndrome: a benign childhood autonomic epilepsy frequently imitating encephalitis, syncope, migraine, sleep disorder, or gastroenteritis. Pediatrics. 2006;118(4):e1237-e1243. https://doi.org/10.1542/peds.2006-0623
- Lada C, Skiadas K, Theodorou V, Loli N, Covanis A. A study of 43 patients with Panayiotopoulos syndrome, a common and benign childhood seizure susceptibility. Epilepsy. 2003;44(1):81-88. https://doi.org/10.1046/j.1528-1157.2003.32602.x
- Panayiotopoulos CP. Autonomic seizures and autonomic status epilepticus peculiar to childhood: diagnosis and management. Epilepsy Behav. 2004;5(3):286-295. https://doi.org/10.1016/j.yebeh.2004.01.013
- Caraballo RH, Sologuestua A, Grañana N, et al. Idiopathic occipital and absence epilepsies appearing in the same children. Pediatr Neurol. 2004;30(1):24-28. https://doi.org/10.1016/S0887-8994(03)00409-0
- Koutroumanidis M. Panayiotopoulos syndrome: an important electroclinical example of benign childhood system epilepsy. Epilepsy. 2007;48(6):1044-1053. https://doi.org/10.1111/j.1528-1167.2007.01096.x
- Girish M, Mujawar N, Dandge V. Panayiotopoulos syndrome. Indian Pediatr. 2008;45(5):420-421.
- Sánchez Fernández I, Loddenkemper T. Pediatric focal epilepsy syndromes: J Clin Neurophysiol. 2012;29(5):425-440. https://doi.org/10.1097/WNP.0b013e31826bd943
- Ozkara Ç, Benbir G, Celik AF. Misdiagnosis due to gastrointestinal symptoms in an adolescent with probable autonomic status epilepticus and Panayiotopoulos syndrome. Epilepsy Behav. 2009; 14(4):703-704. https://doi.org/10.1016/j.yebeh.2009.02.013
- Verrotti A, Sebastiani M, Giordano L, Striano P, Belcastro V, Franzoni E, et al. Panayiotopoulos syndrome with convulsive status epilepticus at the onset: a long-term study. Seizure. 2014;23(9):728-731. https://doi.org/10.1016/j.seizure.2014.05.013
- Oguni H. Treatment of benign focal epilepsies in children: When and how should be treated? Brain Dev. 2011;33(3):207-212. https://doi.org/10.1016/j.braindev.2010.10.024
- Hirano Y, Oguni H, Funatsuka M, Imai K, Osawa M. Neurobehavioral abnormalities may correlate with increased seizure burden in children with Panayiotopoulos syndrome. Pediatr Neurol. 2009;40(6):443-448. https://doi.org/10.1016/j.pediatrneurol.2008.12.005
- Caraballo R, Cersósimo R, Fejerman N. Panayiotopoulos syndrome: a prospective study of 192 patients. Epilepsy. 2007;48(6):1054-1061. https://doi.org/10.1111/j.1528-1167.2007.01085.x
- Sanders S, Rowlinson S, Manidakis I, Ferrie CD, Koutroumanidis M. The contribution of the EEG technologists in the diagnosis of Panayiotopoulos syndrome (susceptibility to early onset benign childhood autonomic seizures). Seizure. 2004;13(8):565-573. https://doi.org/10.1016/j.seizure.2004.01.006
- Parisi P, Villa MP, Pelliccia A, Rollo VC, Chiarelli F, Verrotti A. Panayiotopoulos syndrome: diagnosis and management. Neurol Sci. 2007;28(2):72-79. https://doi.org/10.1007/s10072-007-0790-4
- Martín del Valle F, Díaz Negrillo A, et al. Panayiotopoulos syndrome: probable genetic origin, but not in SCN1A. Eur J Paediatr Neurol. 2011;15(2):155-157. https://doi.org/10.1016/j.ejpn.2010.08.002
- García C, Rubio G. Efficacy and safety of levetiracetam in the treatment of Panayiotopoulos syndrome. Epilepsy Res. 2009;85(2-3):318-320. https://doi.org/10.1016/j.eplepsyres.2009.03.024
- Fitzgerald BJ, Okos AJ, Miller JW. Treatment of out-of-hospital status epilepticus with diazepam rectal gel. Seizure. 2003;12(1):52-55. https://doi.org/10.1016/S105913110200170X
- Glauser T, Shinnar S, Gloss D, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American Epilepsy Society. Epilepsy Curr. 2016;16(1):48-61. https://doi.org/10.5698/1535-7597-16.1.48