
Peer Reviewed
Pulmonary Atresia With Ventricular Septal Defect and Major Aortopulmonary Collateral Arteries
Authors:
Wilson B. Pfeiffer, MS-IV, and Alixandria A. Fiore, MS-IV
Nova Southeastern University, Fort Lauderdale, Florida
Syed A. A. Rizvi, PhD, MS, MBA
Hampton University School of Pharmacy, Hampton, Virginia
Zafar Qureshi, MD
UHI CommunityCare Clinic, Miami, Florida
Daniela Concha-Alecchi, PA-S, and Karen Vejano, PA-S
Keiser University, Fort Lauderdale, Florida
Anthony Centeno, PA-S
Barry University, Miami, Florida
Citation:
Pfeiffer WB, Fiore AA, Rizvi SAA, et al. Pulmonary atresia with ventricular septal defect and major aortopulmonary collateral arteries. Consultant. 2019;59(8):246-248.
A boy was born at 39 weeks of gestation via spontaneous vaginal delivery to a 22-year-old primigravida.
History. The mother had received full prenatal care and had been adherent with all scheduled visits. She denied any family history of genetic disorders. The pregnancy had been uneventful with the exception of maternal chlamydia and bacterial vaginosis, both of which had been treated successfully. Prenatal ultrasonograms at weeks 8, 13, and 20 had been negative for cardiac anomalies and had shown appropriate fetal growth. The delivery had been complicated by meconium-stained amniotic fluid. The nursery course had been complicated by an extended nursery stay of 4 days, which had required treatment with maternal nasal continuous positive-airway pressure.
Physical examination. The newborn’s pulse and activity were normal, with appropriate grimacing present. The Apgar score was 8/8 at 1 and 5 minutes. Oxygen was given via facemask, which elicited a poor response. Maximum preductal oxygen saturation was 90%. There was a grade 3/6 holosystolic murmur heard upon auscultation on the left sternal border. The infant was placed on 2 L of 100% oxygen via nasal cannula, was started on prostaglandin E1 at 0.01 µg/kg/min, and then was admitted to the neonatal intensive care unit for further monitoring and testing.
Diagnostic tests. Laboratory workup findings included the following values: white blood cell count, 11.8 x 103/µL (reference range, 4.0-11.0 x 103/μL); hemoglobin, 17.2 g/dL (reference range, 13.8-17.2 g/dL); hematocrit, 52.0% (reference range, 40.0%-52.0%); nonfasting plasma glucose, 112 mg/dL (reference range, 90-125 mg/dL); sodium, 140 mEq/L (reference range, 135-146 mEq/L); potassium, 3.4 mEq/L (reference range, 3.8-5.1 mEq/L); calcium, 9.2 mg/dL (reference range, 9.2-10.0 mg/dL); creatinine, 0.74 mg/dL (reference range, 0.7-1.3 mg/dL); chloride, 106 mEq/L (reference range, 96-106 mEq/L); total bilirubin, 9.7 mg/dL (reference value, ≤5.1 mg/dL); and direct bilirubin, 0.11 mg/dL (reference value, ≤0.3 mg/dL).
Echocardiography findings showed pulmonary atresia with ventricular septal defect (PA-VSD) and major aortopulmonary collateral arteries (MAPCAs) (Figure 1). The interatrial septum was floppy and aneurysmal with a large secundum atrial septal defect (ASD) with predominantly left-to-right shunting. Mild tricuspid regurgitation was noted, with a possible association to small left ventricle to right atrium shunt, as well as mild right ventricle enlargement and normal left ventricle size. The pulmonary valve was atretic with hypoplastic, confluent main and branch pulmonary arteries, fed by multiple aortopulmonary collaterals. Furthermore, indeterminate arch sidedness with large collaterals was seen coming off the descending aorta.
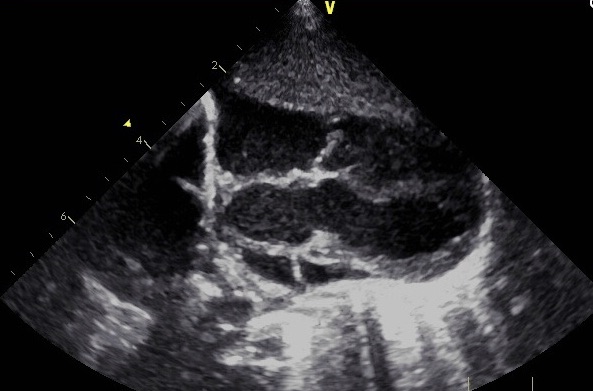
Figure 1. Echocardiogram showing the presence of PA-VSD.
Computed tomography (CT) arteriography of the chest showed levocardia and levoposition. There was enlargement of the right ventricle and right atrium. The left ventricle and left atrium were normal in size. Ventricular septal defect was seen, with a mild overriding right-sided aorta. A left innominate artery was present, which gave rise to the left subclavian and carotid artery. There was a separate origin of the right common carotid artery and right subclavian artery.
Along the proximal descending aorta, there was a large collateral originating from the left anterior aspect of the descending aorta, which gave rise to vessels that extended to the left lower lung (Figure 2). There were small branches that extended to the left upper lung as well as the right upper lung. There were tortuous branches that coalesced with the right main pulmonary artery branch. There was severe narrowing of the origin of the left main branch pulmonary artery.
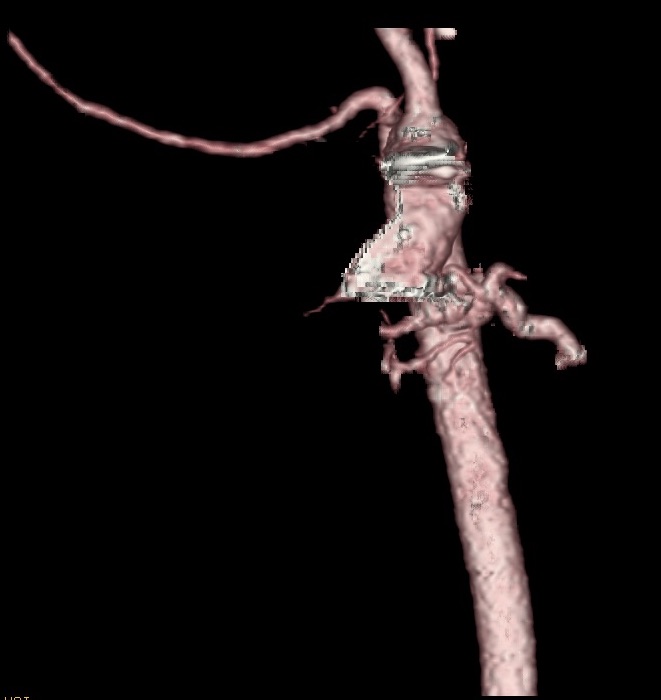
Figure 2. CT arteriogram showing the presence of major collateral arteries branching from both the distal ascending and proximal descending aorta.
T-cell flow cytometry assay showed the following: CD3 cells, 44.58% (low); absolute CD3 cell count, 841/µL (reference range, 2500-5500/µL); CD4 cells, 32.15% (low); absolute CD4 cell count, 600/µL (reference range, 1600-4000/µL); absolute CD8 cell count, 231/µL (reference range, 560-1700/µL); CD19 cells, 36.3% (high); absolute CD19 cell count, 692/µL (reference range, 300-2000/µL); CD56 cells, 17.3% (normal); and absolute CD56 cell count, 332/µL (170-1100/µL).
Genetic test results showed a deletion at 22q11.2. Results of flow cytometry and genetic testing were consistent with DiGeorge Syndrome.1
Discussion. PA-VSD and MAPCAs is a complex cardiac lesion that is extremely rare.2 The Baltimore-Washington Infant Study reported a PA-VSD incidence of 0.07 per 1000 live births, accounting for only 1.5% of all forms of congenital heart disease. Of all babies born with PA-VSD, only 25% are found to also have MAPCAs.3
The lesion is defined by the malformation of the pulmonary valve with a patent defect in the interventricular septum, with oxygenation occurring by means of major collateral arteries branching off of the thoracic aorta. Most commonly, collaterals will branch from the descending aorta; however, a few cases have been reported in which the ascending aorta becomes a branch point.4 Diagnostic evaluation includes echocardiography and CT angiography, with the goal being to determine the intrinsic cardiac anatomy as well as the presence or absence of collateral arteries.5 In noncyanotic neonates, another goal of diagnostic radiology is to determine the source of oxygenation—most importantly, whether or not the neonate is dependent upon a patent ductus arteriosus for oxygenation. Until receiving confirmation that the newborn is not ductal-dependent for adequate oxygenation, prostaglandin E1 must be administered for the maintenance of the ductus arteriosus.6
Our patient’s case was unique in several ways. Fewer than 1 in 14,000 children are born with PA-VSD. Of that group, only 1 of 4 will also have MAPCAs as their means of oxygenation. Of the neonates with MAPCAs, most will have collaterals branching off of the descending aorta.7
In our patient’s case, not only was the boy born with PA-VSD, but he also had MAPCAs as his means of oxygenation; furthermore, he had collaterals branching off of both the ascending and descending aorta. Of even more importance is the fact that there were no prenatal screening tests that predicted the presence of a cardiac anomaly, including prenatal ultrasonography.
This case illustrates the dire importance of neonatologists to be diligent when evaluating newborns, even when no abnormalities are expected. In our patient’s case, the only abnormality seen on delivery was tachypnea and noticeable pallor; there was no cyanosis and no respiratory distress. However, due to the tachypnea and pallor, echocardiography was done to assess for cardiac lesions, only to find one of the rarest conditions known of cardiac origin, of which requires serious medical attention.
Definitive treatment of PA-VSD with MAPCAs requires surgery, with the goal being the creation of separate pulmonic and systemic circulations, which requires the closure of the VSD and the recreation of the pulmonary valve outflow track.8
Outcome of the case. Our patient was stabilized in the neonatal intensive care unit and was sent home on day 3 after multiple reassuring test results were obtained. Cardiac catheterization was scheduled for week 6, with reconstructive cardiac surgery to follow. Most recently, the infant was being treated symptomatically for DiGeorge syndrome as we pursue the definitive treatment in the future.
- Pierdominici M, Marziali M, Giovannetti A, et al. T cell receptor repertoire and function in patients with DiGeorge syndrome and velocardiofacial syndrome. Clin Exp Immunol. 2000;121(1):127-132.
- Presnell LB, Blankenship A, Cheatham SL, Owens GE, Staveski SL. An overview of pulmonary atresia and major aortopulmonary collateral arteries. World J Pediatr Congenit Heart Surg. 2015;6(4):630-639.
- Ozden K, Mutlu B, Kahveci G, et al. Pulmonary atresia and ventricular septal defect with MAPCAs associated with right sided endocarditis and paradoxical embolic event. Eur J Echocardiogr. 2007;8(4):292-295.
- DeRuiter MC, Gittenberger-de Groot AC, Poelmann RE, Vanlperen L, Mentink MMT. Development of the pharyngeal arch system related to the pulmonary and bronchial vessels in the avian embryo: with a concept on systemic-pulmonary collateral artery formation. Circulation. 1993;87(4):1306-1319.
- Nikolaou K, Alkadhi H, Bamberg F, Leschka S, Wintersperger BJ. MRI and CT in the diagnosis of coronary artery disease: indications and applications. Insights Imaging. 2011;2(1):9-24.
- Yun SW. Congenital heart disease in the newborn requiring early intervention. Korean J Pediatr. 2011;54(5):183-191.
- Mun D-N, Park CS, Kim Y-H, Goo HW. Pulmonary atresia with ventricular septal defect and major aortopulmonary collaterals associated with left pulmonary artery interruption. Korean J Thorac Cardiovasc Surg. 2016;49(5):374-378.
- Lofland GK. The management of pulmonary atresia, ventricular septal defect, and multiple aorta pulmonary collateral arteries by definitive single stage repair in early infancy. Eur J Cardiothorac Surg. 2000;18(4):480-486.